|
Hochdruck-Flüssigkeitschromatographie
(HPLC) HPLC steht für High Pressure
Liquid Chromatography (manche sprechen auch von High Performance Liquid Chromatography).
Die HPLC ist eine sehr leistungsfähige chromatographische Technik zur Auftrennung und
Analyse von Stoffgemischen. Sie gehört zur Gruppe der Säulen-Chromatographien, die
stationäre Phase ist in eine Stahlsäule gepackt ist. Flüssigkeiten bilden die mobile
Phase.
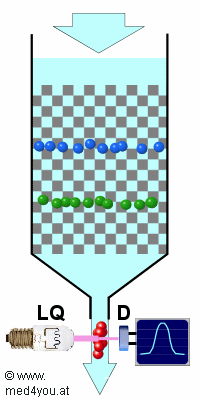 |
Das Prinzip ist nicht außergewöhnlich
Das Prinzip der Auftrennung bei der HPLC gleicht anderen Chromatographieverfahren: Die zu
analysierende Probe (farbige Kugeln) wird von einer durch die Säule fließende
Flüssigkeit (=die mobile Phase) mitgenommen.
Die einzelnen Bestandteile der Probe wandern aber ungleich schnell, weil sie durch
Wechselwirkungen mit der stationären Phase (grau) unterschiedlich stark aufgehalten
werden.Beim Austritt aus der Säule kann man die einzelnen
Stoffe mit einem geeigneten Detektor (D) nachweisen. Das ist aber auch nichts
besonderes, das macht man bei der normalen Säulen-Chromatographie auch. |
Was ist das Besondere an der HPLC?
Wie der Name sagt, ist das der hohe Druck, mit dem man die mobile
Phase durch die Säule pumpt. Aber der Druck ist natürlich nicht Selbstzweck:
Das Problem: Will man bei der
Säulen-Chromatographie eine gute Auftrennung, braucht man eine lange Säule und eine
möglichst kleine Partikelgröße der stationären Phase. Aber diese Faktoren verlangsamen
die Analyse beträchtlich. Man muss Kompromisse schließen, damit die Flüssigkeit rasch
genug durch die Säule fließt. Sie wird ja nur von der Schwerkraft angetrieben. Die
Trennleistung der normalen Säulen-Chromatographie war daher für viele Anwendungen nicht
ausreichend.
Der Ausweg: man setzt Druck ein, um die
Flüssigkeit durch die Säule zu pumpen. Dann kann man viel kleinere Partikel
als stationäre Phase einsetzen, hat dadurch eine hervorragende Trennung
und kann relativ kurze Säulen einsetzen. Außerdem läuft die Trennung
schneller ab.
In Zahlen heißt das:
- Normale Säulen-Chromatographie: Partikelgrößen meist von
100 - 300 µm Durchmesser, Säulenlänge von 0.5 - 3 m,
Säulendurchmesser 0.5 - 5 cm.
- HPLC: Partikelgrößen meist 3 - 10 µm, Säulenlänge
5 - 30 cm, Säulendurchmesser (innen) 2 - 4 mm.
 |
Typische HPLC-Säule aus Stahl |
Man braucht dazu allerdings relativ hohe Drücke. Zur Verwendung
einer HPLC-Säule kann ein Druck von 100 - 200 bar notwendig werden.
UHPLC hat noch höhere Trennleistung
In Entwicklung sind HPLC-Systeme, die Säulen mit noch kleineren
Partikeln verwenden (1 µm) und daher noch höhere Drücke (2000 bar und mehr!)
einsetzen müssen: Ultrahigh-Pressure Liquid Chromatography (UHPLC).
Aufbau eines HPLC-Systems
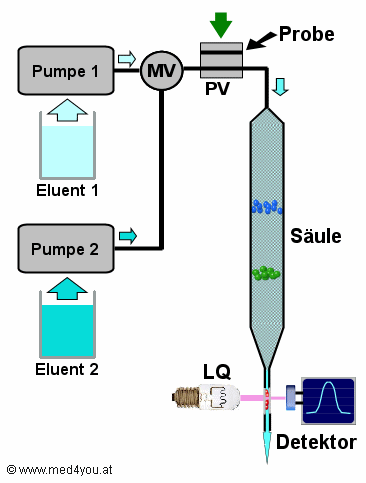 |
Einfacher als es aussieht
Vergessen wir vorerst die 2. Pumpe, das Mischventil MV und das Probenventil
PV. Dann ist es ganz einfach: Pumpe 1 pumpt die mobile Phase (den Eluenten) durch die
Säule. Der nimmt die verschiedenen Stoffe der Probe verschieden schnell mit. Beim
Austritt werden die Stoffe gemessen.
Wie bringt man eine Probe in so ein Hochdrucksystem?
Man gibt sie in aller Ruhe mit einer Spritze in den oberen Kanal des Probenventils
PV (schwarzer Pfeil) und schiebt das PV rasch nach unten (grüner Pfeil). Die Probe wird
dann von der mobilen Phase zur Säule transportiert. |
| |
|
| Vom Schema zur Realität: |
|
 |
HPLC-Kompaktsystem
So sieht ein kompaktes HPLC-System aus.
Man erkennt aber nicht viel. Außer die Behälter für die Eluenten (=mobile Phase) links
oben und den Auswerte-Computer rechts.
(Photo der Firma Shimadzu) |
Neben den Kompaktsystemen kann man auch einzelne Module kaufen
und zusammenstellen.
 |
Pumpe eines Modulsystems
Die Pfeile zeigen auf das typische "Schlauchsystem" einer HPLC: Wegen des hohen
Drucks müssen Stahlrohre verwendet werden (sind aber biegsam).
(Photo der Firma Shimadzu) |
Wie sieht das Ergebnis einer HPLC aus?
Man misst mit dem Detektor (das könnte ein einfaches Photometer sein) kontinuierlich am Ende der Säule. Die
Messwerte stellt man als Kurve dar und erhält dadurch das Chromatogramm. Die einzelnen
Erhebungen nennt man Peaks.
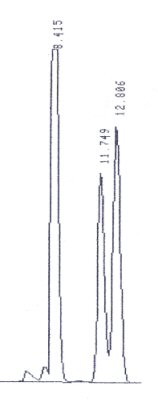 |
Trennung von Zuckern
So könnte ein kurzes Chromatogramm aussehen. Man sieht drei dominierende Peaks. In diesem
Beispiel die Zucker Maltose, Rhamnose und Fucose. Woher man das weiß? Weil das keine
echte Probe ist, sondern ein Gemisch aus diesen drei Zuckern. Woher man weiß, welcher
welcher ist? Man analysiert die Zucker vorher einzeln.
Hat man das einmal ausgetestet, kann man Proben laufen lassen. Findet man dann einen Peak
an der Stelle, an der normalerweise Stoff X kommt, dann kann es sich auch in der Probe um
diesen Stoff X handeln.
Natürlich nur, wenn kein anderer Stoff dabei stört und "darunter
liegt". Das muss man vorher durch geeignete Trennungsbedingungen, durch Wahl des
passenden Detektors und vielleicht auch durch spezielle Vor- oder Nachbehandlung der Probe
sicherstellen.
|
Die Zahlen oberhalb der Peaks sind die sog. Retentionszeiten
(in Minuten). Das ist die Zeit vom Start der Analyse bis zur Detektion des Peaks. Und die
sind wichtig: denn jetzt weiß man, wann welcher Stoff kommen sollte. Und das sollte bei
gleichbleibenden Bedingungen (und für diese muss man sorgen) gleich bleiben. Man muss
einen Stoff an der Retentionszeit erkennen, ein Peak schaut ja wie der andere aus.
Die Fläche unter den Peaks ist proportional der Konzentration des
Stoffs. Die HPLC liefert dadurch sehr präzise quantitative Ergebnisse.
Wozu ist jetzt die 2. Pumpe gut?
Kommen wir auf die Abbildung zurück. Da war eine 2. Pumpe und ein
Mischventil eingezeichnet. Zur Erklärung, warum man dies braucht, hilft folgende
Animation.
 |
Die blauen Kugeln brauchen zu lange
Beachten wir nur die Wanderungsgeschwindigkeit der verschiedenen Stoffe. Die roten Kugeln
laufen sehr schnell und kommen bald aus der Säule. Die grünen schon langsamer. Und die
blauen wandern extrem langsam. |
Das ist aus verschiedenen Gründen unbefriedigend:
- Es dauert zu lange bis man die blauen messen kann.
- Es ist auch nicht gut, wenn Stoffe zu lange in der Säule wandern.
Die Peaks werden dann breiter und ein Peak könnte mit einem Nachbarpeak verschmelzen.
D.h., die Trennung wird schlechter.
1. Lösungsansatz: Man nimmt eine mobile Phase, die
die Stoffe schneller aus der Säule wäscht. Das ist nicht schwer. Die blauen Kugeln
werden dann schneller kommen und schönere Peaks machen. Aber die ersten Stoffe werden
vielleicht zu schnell kommen. Die roten und die grünen Kugeln könnten gleichzeitig
kommen. Will man alle Stoffe analysieren, ist das daher keine gute Lösung. Will man nur
die blauen, könnte man das so machen.
Besserer Lösungsansatz: Man nimmt 2 oder mehrere
mobile Phasen (=Eluenten). Man setzt zuerst einmal die "milde" mobile Phase ein,
wartet bis die roten und die grünen Kugeln aus der Säule sind. Dann erst nimmt man die
"scharfe" mobile Phase und erreicht so, dass auch die blauen Kugeln rasch und
mit einem halbwegs schlanken Peak aus der Säule kommen.
So erklären sich die 2 eingezeichneten Pumpen, Eluenten und das
Mischventil. In der Praxis wechselt man meist nicht plötzlich von Eluent 1 auf
Eluent 2 sondern die Pumpen oder das Mischventil werden so programmiert, dass dem
Eluenten 1 immer mehr Eluent 2 beigemischt wird. Man mischt also kontinuierlich oder
in Stufen immer mehr Eluent 2 (und ev. Eluent 3) dazu. Dies nennt man Gradient
oder auch Gradiententrennung. Für viele Anwendung ist eine solche
Gradiententrennung notwendig.
Für einfachere Aufgaben braucht man dies nicht. Dann genügt ein Eluent. Dies nennt man isokratische
Trennung.
Säulen
Säulen in der HPLC besitzen normalerweise einen Stahlmantel und
sind mit einem Material mit sehr niedriger Teilchengröße (3 - 10 µm)
befüllt. Manchmal dient die ursprüngliche Teilchenoberfläche als stationäre Phase. Oft
werden die Teilchen chemisch mit einer Schicht überzogen, die dann die stationäre Phase
bildet. Je nach gewünschter Anwendung bindet die Oberfläche der Partikel eher
wasserlösliche Substanzen, wasserunlösliche Substanzen oder dient als Ionen-Austauscher.
Es gibt eine Unzahl von verschiedenen Materialien für die unterschiedlichsten
Anwendungen.
Reversed-Phase (RP) Chromatographie: Erwähnt soll noch der Ausdruck
"RP-Säule" werden: Normalerweise trägt die Oberfläche des Füllmaterials
(=stationäre Phase) polare* Gruppen. Und als Eluent verwendet man apolare (oder wenig
polare) Flüssigkeiten.
Das kann man umdrehen: Man kann die Oberfläche der stationären Phase mit langen
Kohlenstoffketten überziehen. Dann wird sie apolar und man nimmt eher polare
Flüssigkeiten als mobile Phase. Die Phasen sind umgedreht (reversed). Daher der Ausdruck
"Reversed-Phase" oder Reversed-Phase Säule, kurz RP-Säule.
*Die Erklärung der Begriffe polar/apolar würde zu tief in die Chemie bzw. Physik
führen. Keine Erklärung aber praktisch wichtig: Polare Stoffe lösen sich gut in polaren
Flüssigkeiten (z.B. Wasser), apolare in apolaren (z.B. Benzin, Hexan)
Vorsäulen (Guard-Columns = Schutzsäulen)
erhöhen die Lebensdauer der Säule
Verunreinigungen in der Probe können die Lebensdauer eine Säule
beträchtlich verkürzen. Billigere Säulen tauscht man einfach regelmäßg aus. Bei
teureren kann man vor die eigentliche Säule eine billige, kurze Vorsäule schalten. Sie
enthält das gleiche oder ein ähnliches Packungsmaterial wie die Hauptsäule und fängt
so die Verunreinigungen der Probe ab. Man muss sie aber regelmäßig tauschen.
Detektoren
Der Detektor wird nach der Säule angebracht und beobachtet die
austretende mobile Phase, in der die Stoffe einer nach dem anderen herauskommen.
- Der Detektor kann wie ein Photometer arbeiten und die Abschwächung
eines Lichtstrahls detektieren (im sichtbaren oder im UV Bereich): UV/Visible-Detektor.
- Fluoreszenzdetektoren messen die durch einen
Anregungsstrahl ausgelöste Fluoreszenz, um fluoreszierende Stoffe zu erkennen.
- Man kann aber auch Änderungen der Lichtbrechung detektieren: Brechungsindex-Detektor
(Refractive-Index bzw. RI-Detektor).
- Elektrochemische Detektoren (ECD) detektieren
Änderungen des Stromflusses zwischen zwei Polen, der durch die austretenden Stoffe
verursacht wird.
- Dioden-Array Detektoren
Man kann eine Substanz mit einem UV/Visible-Detektor, also mit einer Art Photometer, bei
einer bestimmte Wellenlänge detektieren. Man kann es aber auch bei 2 Wellenlängen
machen. Man erhält so zusätzliche Information. Noch mehr Information erhält man, wenn
man bei sehr vielen Wellenlängen misst. Man bekommt dann zu jedem Peak eine Art
Spektralkurve. Das hilft, Substanzen und Störsubstanzen zu erkennen, denn man kennt das
Spektrum der Substanz, die man erwartet. Ist das plötzlich verändert, könnte eine
andere Substanz darunter liegen.
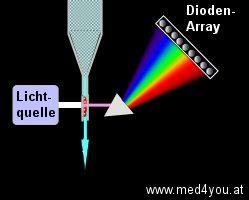 |
Funktionsweise: Der Lichtstrahl tritt durch die mobile Phase
und durch die darin gelösten Stoffe, wird von einem Gitter oder einem Prisma in seine
Spektralfarben zerlegt und trifft dann auf eine Reihe von lichtempfindlichen Dioden, den
Dioden-Array. |
Post-Column-Derivatisierung
(NACHsäulenderivatisierung)
Nicht alle Stoffe kann man in einem Detektor auf einfache Weise
nachweisen. Manche muss man dazu erst mit einem anderen Stoff reagieren lassen. Das tut
man, indem man der mobilen Phase, nachdem sie aus der Säule ausgetreten ist, z.B. einen
Farbstoff beimischt. Erst dann geht es weiter zum Detektor.
Das klassische Beispiel ist die Post-Column-Färbung von Aminosäuren mit
Ninhydrin.
Pre-Column-Derivatisierung (VORsäulenderivatisierung)
Bei der Post-Column-Derivatisierung werden die Substanzen nach der
Auftrennung, also nachdem sie die Säule verlassen haben, verändert (z.B. gefärbt). Das
ist ein nicht geringer apparativer Aufwand. Man braucht eine eigene Pumpe, ein T-Stück
zum Mischen von mobiler Phase und Farbstoff. Das ganze muss eventuell auch in einigen
Spiralenwindungen durch einen Ofen fließen, damit die Reaktion stattfindet.
In manchen Fällen kann man die interessierenden Stoffe schon vor der Trennung färben.
Alle auf einmal im Probenfläschen. Erst dann wird die Probe analysiert. Man nennt dies
Pre-Column-Derivatisierung.
Ein Beispiel wäre die Orthophthal-Aldehyd (=OPA)-Methode zur
Aminosäurenbestimmung. OPA ist ein fluoreszierender Farbstoff. Diesen koppelt man vor der
Auftrennung an die Aminosäuren. Man kann sie dann nach der Auftrennung mit einem
Fluoreszenzdetektor nachweisen.
Externer und interner
Standard
Es wurde schon erwähnt: Die Fläche unter dem Peak ist proportional
zur Konzentration. Aber wie erhält man wirklich quantitative Ergebnisse, wie berechnet
man die Konzentration einer Substanz?
Da gibt es 2 prinzipielle Möglichkeiten.
- Der externe Standard
Man stellt eine Lösung mit einer bestimmten Konzentration des interessierenden Stoffs
her. Diese Standard-Lösung lässt man laufen. Man wird einen Peak erhalten und ein
bestimmtes Verhältnis zwischen Peakfläche und Konzentration errechnen können. Diesen
Faktor verwendet man für die Peaks in den zu messenden Proben.
Das funktioniert aber nur unter gewissen Voraussetzungen: Man muss immer exakt die gleiche
Probenmenge aufgeben. Die Bedingungen z.B. bei Derivatisierungen müssen immer exakt
gleich bleiben. Und auch alle anderen Bedingungen sollten sich nicht ändern, sie könnten
sonst das Ergebnis beeinflussen.
Man kann mit externem Standard arbeiten, es funktioniert. Aber es gibt eine sicherere
Methode.
- Der interne Standard
Dabei gibt man zu der zu analysierenden Probe eine bekannte, den zu messenden Substanzen
ähnliche Substanz hinzu. Natürlich eine bestimmte Menge. Diese Substanz dient als
interner Standard, sie ist in der Probe. Ab dem Zeitpunkt der Zugabe bis
zur Messung macht der interne Standard alles mit, was auch die zu messenden Substanzen
mitmachen. Man wertet den internen Standard (der ein schöner, gut getrennter Peak sein
sollte) gemeinsam mit den anderen aus und berechnet die Konzentrationen der zu messenden
Substanzen in Relation zum internen Standard.
Vorteile: Hat man versehentlich etwas weniger Probe aufgegeben oder ist einmal die
Post-Column-Färbung etwas schwächer ausgefallen. Kein Problem, auch der interne Standard
wird ein kleinerer Peak werden. Im Verhältnis wird es passen und die Ergebnisse werden
korrekt sein. Arbeitet man mit automatischen Probengebern über Nacht, in denen die
letzten Proben vielleicht 16h nach den ersten gemessen werden, dann könnten diese durch
Verdunstung konzentriert worden sein. Macht nichts, auch der interne Standard wird
konzentrierter sein. All diese Probleme würden bei Einsatz eines externen Standards zu
Fehlern führen.
Anwendungen in der Medizin
Im Routinelabor eher selten eingesetzt, eventuell zur Bestimmung des
HbA1C. In etwas spezialisierteren
Labors analysiert man damit Hämoglobine (bei Verdacht auf erbliche
Hämoglobin-Fehlbildungen wie z.B. Thalassämie), Aminosäuren (Vd.a.
Stoffwechselkrankheiten), Vitamine, Medikamente, Drogen, Catecholamine und deren
Metaboliten (Blutdruckprobleme), andere Hormone, Hydroxyindolessigsäure (Vd.a.
Karzinoidsyndrom), Porphyrine (Vd.a. Porphyrinurie).
|
|
|
Gaschromatographie
(GC) Die Gaschromatographie ist eine
Methode zur Auftrennung und Analyse von Stoffgemischen. Wie bei anderen
chromatographischen Verfahren ist die Basis der Auftrennung die unterschiedliche
Verteilung der Stoffe zwischen einer stationären und einer mobilen Phase. Während sich
die mobilen Phase an der stationären Phase vorbei bewegt, nimmt sie die Stoffe mehr oder
weniger schnell mit. Stoffe, die sich stark an die stationäre Phase binden, werden
langsam mitwandern, solche die mehr Affinität zur mobilen Phase haben, werden schneller
mitwandern.
Mobile Phase
Das Besondere an der Gaschromatographie ist, dass die mobile Phase ein Gas und keine
Flüssigkeit ist. Daher müssen die zu analysierenden Stoffe verdampfbar sein oder in
verdampfbare Verbindungen überführbar sein.
Stationäre Phase
Die stationäre Phase (Flüssigkeiten oder Festsubstanzen) der GC befindet sich in einer
relativ langen, spiralig angeordneten Säule. Diese ist entweder mit der stationären
Phase bepackt oder ihre Innenwand wird von der stationären Phase ausgekleidet.
Aufbau eines Gaschromatographen
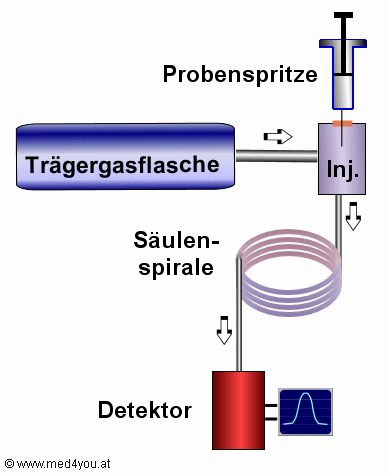 |
Mit der Spritze bringt man die Probe in den Injektor. Dort wird sie
unter Wärmeeinfluss verdampft. Der Gasstrom aus der Trägergasflasche nimmt die Probe zur
Säule mit. In der Säule werden die Stoffe aufgetrennt: manche wandern schneller, manche
langsamer durch die Säule.
Der Detektor zeichnet das Austreten der Stoffe auf.
Ergänzung: man kann flüssige Proben auch direkt auf die Säule aufgeben. Die
Probe verdampft dann in der Säule. |
Ein GC System in der Realität
 |
Gaschromatograph
Bei diesem Kompaktsystem kann man die einzelnen Bausteine kaum erkennen. Die Tastatur zur
Steuerung sieht man unten links. Der Regler für die Kontrolle der Gasströmungen
fällt rechts oben auf.
Dieser GC braucht nicht viel Platz. Dazu kommt aber noch mindestens eine Gasflasche.
(Photo: Fa. Shimadzu) |
Wie sieht das Ergebnis einer GC aus?
Man misst mit dem Detektor kontinuierlich am Ende der Säule. Die
Messwerte stellt man als Kurve dar und erhält dadurch das Chromatogramm. Die einzelnen
Erhebungen nennt man Peaks.
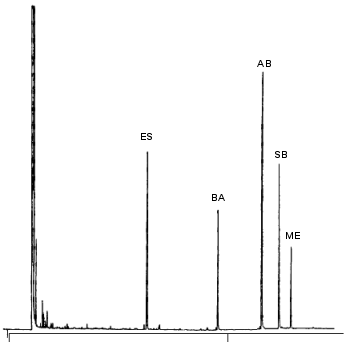 |
Trennung von Medikamenten
Gaschromatographische Trennung von Medikamenten (Ethosuximid und verschiedene
Baribituraten). Wie bei der HPLC ist die Zeit, die eine Substanz braucht, bis sie aus der
Säule kommt, wichtig. Anhand dieser Zeit kann man erkennen, um welche Substanz es sich
handelt.
Und wie bei der HPLC klärt man vorher durch Analyse von Einzelstandards, wann
welche Substanz kommt.
|
Säulen
Man unterscheidet vor allem 2 Arten von Säulen
- Gepackte Säulen (Packed Columns)
Gepackte Säulen sind Glas oder Stahlrohre (innerer Durchmesser meist 1-5 mm), in die
die stationäre Phase (Pulver kleiner Teilchen von 100-300 µm) gepackt wurde. Diese
Säulen können einige Meter lang sein (werden zur Spirale geformt).
Vorteil: verträgt größere Probenmengen. Nachteil: geringe Trennleistung.
Trennmechanismus
Die Teilchen in der Säule (z.B. Aluminiumoxidpulver) kann man als solche zur Trennung
verwenden. Man nennt das auch GSC, Gas-Solid-Chromatography (übersetzt:
Gas-Feststoff-Chromatographie). Dann basiert die Trennung vorwiegend auf Adsorptionsvorgängen. Oder man
überzieht die Teilchen mit einer Flüssigkeit. Das nennt man GLC,
Gas-Liquid-Chromatography (übersetzt: Gas-Flüssigkeits-Chromatographie). Dann liegen der
Trennung vor allem Verteilungsvorgänge
zu Grunde.
- Kapillarsäulen (Capillary Columns)
 |
Kapillarsäulen sind dünne Kapillaren aus Glas-ähnlichem
Material. Der innere Durchmesser ist nur 0.1-0.5 mm. Die Länge liegt meist zwischen
10 und 150 m.
(Photo: Fa. Kemomed) |
In Kapillarsäulen wird keine stationäre Phase gepackt, sie
bilden eine sehr dünne Röhre, eine Kapillare. Wo ist die stationäre Phase? Die kleidet
die Wand der Kapillare aus. Meist ist dies eine Flüssigkeit, die an der Innenwand einen
Film bildet. Die Säulen gibt es fertig mit Flüssigkeitsauskleidung zu kaufen.
Bei Flüssigkeit darf man sich nichts Wasser-ähnliches vorstellen. Die
Flüssigkeiten bei der GC sind dickflüssig und schwer verdunstend. Verwendet werden z.B.
Silikonpolymere oder Silikonpolyester.
Kapillarsäulen zeigen eine hervorragende Trennleistung. Nachteil: es dürfen nur sehr
kleine Probenmengen auf die Säule aufgegeben werden.
Trennmechanismus
Bei Kapillarsäulen mit Flüssigkeitsauskleidung erfolgt die Trennung der Stoffe durch Verteilungsvorgänge zwischen einem
Gas und einer Flüssigkeit (GLC, Gas-Liquid-Chromatography).
Es gibt noch andere Typen von Kapillarsäulen, diese werden aber seltener
eingesetzt.
Mobile Phasen (Gase)
Als mobile Phase eignen sich z.B. Stickstoff, Helium oder Argon. Sie
müssen von großer Reinheit sein, da Verunreinigungen die Säule schädigen können.
Der Gasfluss hängt von der Säule ab. Während gepackte Säulen eine Flussrate von 10 bis
60 ml/min brauchen, fließen durch Kapillarsäulen nur etwa 1 - 2 ml/min.
Der Gasfluss muss genau kontrolliert* sein, er ist für die Trennung sehr wichtig.
*Im einfachsten Fall muss der Fluss immer konstant gehalten werden. Für
speziellere Anwendungen (z.B. programmierte Änderungen der Säulentemperatur während der
Trennung) darf er sich auch während Analyse ändern, aber immer unter kontrollierten,
gleichbleibenden Bedingungen (2 Methoden: man hält den Druck konstant oder den Fluss).
Injektor (Probengeber)
Für die meisten Anwendungen werden die Proben in Lösungsmitteln
gelöst und dann mit einer Spritze in das Gerät aufgegeben. Mit speziellen Spritzen oder
speziellen Probengebern kann man auch Gase aufgeben.
Es gibt einige Methoden zur Probenaufgabe. Zwei prinzipiell unterschiedliche seien
herausgegriffen:
- Herkömmliche Methode
Man sticht mit der probengefüllten Spritze durch eine hitzebeständige Membran und bringt
die Probe in einen beheizten Bereich des Injektors ein (und in den Trägergasstrom). Die
Probe verdampft durch die Hitzeeinwirkung und wird vom Trägergas in die Säule getragen.
- On-Column-Methode ("Auf-Säulen"-Methode)
Die dünne Nadel der Spritze wird bis zur Säule geführt. Die Probe wird (noch in
flüssiger Form) in die Säule aufgegeben. Die Verdampfung findet in der Säule statt.
Diese Art der Probenaufgabe hat stark an Bedeutung und Verbreitung gewonnen.
Der Säulenofen
Bei der HPLC braucht man ihn nicht für alle Anwendungen, bei der GC
ist er von größter Wichtigkeit: der Säulenofen. Die Temperatur der Säule muss bei der
GC genauestens eingehalten werden. Sie hat großen Einfluss auf die Trennung und die
Ergebnisse. Auch setzt man häufig Temperaturprogramme ein. Darunter versteht man
programmierte Änderung der Säulentemperatur während der Analyse.
Detektoren
Es gibt eine Vielzahl von Detektoren für die GC, weiter verbreitet
sind:
- Wärmeleitungsdetektor (TCD, Thermal Conductance Detector)
Die Wärmeleitfähigkeit des aus der Säule austretenden Trägergases ändert sich durch
die Beimengung der austretenden Stoffe. Das kann mit geeigneten Apparaturen gemessen
werden.
- Flammen-Ionisationsdetektor (FID)
Die aus der Säule kommende Stoffe werden mit Wasserstoff und Luft vermischt und
verbrannt. Dabei entstehen geladene Teilchen (Ionen) die mit einer Elektrode (also
"elektrisch") gemessen werden.
Die Ionen erhöhen die Leitfähigkeit der Flamme. Der Strom nimmt zu.
- Flammenphotometrischer Detektor (FPD)
Die aus der Säule kommenden Stoffe werden mit Wasserstoff und Luft verbrannt. Dabei
senden Phosphor- oder Schwefel-haltige Verbindungen Licht ganz bestimmter Wellenlänge
aus. Dieses Licht kann man messen.
- Electron Capture Detektor (ECD, Elektronenen-Einfang)
Durch die Wirkung eines radioaktiven Stoffs entstehen im Trägergas Elektronen, die einen
Stromfluss zwischen zwei Elektroden ermöglichen, den man messen kann. Kommen Stoffe aus
der Säule, die diese Elektronen einfangen, nimmt der Stromfluss ab.
Funktioniert nur für Stoffe, die Elektronen einfangen können oder die man
chemisch so verändern kann, dass sie dies tun.
Quantitative Auswertung nach der Peakfläche
Wie bei der HPLC ist auch bei der GC die Fläche des Peaks
proportional zur Stoffmenge. Über externen und internen Standard siehe entsprechenden Absatz bei der HPLC.
Die GC-MS (GC-Massenspektrometrie) erlaubt
eine eindeutige Identifizierung von Stoffen
In der GC entstehen aus dem Stoffgemisch einzelne Stoffe bzw. Peaks.
Um diese näher zu untersuchen, wird jeder Stoff bzw. Peak in der sog. Massenspektrometrie
weiter analysiert. Das Ergebnis ist ein Massenspektrogramm. Während Peaks alle ziemlich
gleich aussehen, ist das Massenspektrogramm jedes Stoffs charakteristisch. Dadurch
lassen sich Stoffe meist eindeutig definieren.
Alle Peaks eines GC-Chromatogramms sehen ziemlich gleich aus. Nur die
Retentionszeit des Peaks, also die Zeitspanne zwischen Aufgabe der Probe und Detektion des
Peaks, sagt uns, um welchen Stoff es sich handelt. Oder besser: handeln könnte. Es ist
nie 100%ig auszuschließen, dass ein anderer Stoff, mit dem man nicht gerechnet hat, an
derselben Stelle im Chromatogramm auftaucht. Hat das Ergebnis rechtliche Konsequenzen
(Drogennachweis), kann eine GC-Analyse allein zu wenig sein. Eine anderes Problem sind
Peaks, die man überhaupt nicht zuordnen kann.
Um einen Peak in der GC näher zu untersuchen kann man ein Massenspektrometer
anschließen. Das wird gleich ans Ende des GC, also nach der Säule angeschlossen. Das
Massenspektrometer zerlegt den Stoff in viele verschiedene geladene Teilchen (Ionen) und
analysiert die entstandenen Teilchen. Genauer gesagt, misst es das
Masse/Ladungs-Verhältnis aller entstandenen Teilchen. Und für viele Stoffe gibt es da
ein bestimmtes Muster, an dem man sie erkennen kann.
Anwendungen in der Medizin
Einsatzmöglichkeiten in der klinischen Diagnostik:
- Hormonbestimmungen
- Vitamine
- Manche Spurenelemente
- Medikamente
- Drogen
- Alkohol
- andere Gifte
|
|

